FDA Sets Safety Standards for Genome Editing Therapies, Building Path Toward Personalized CRISPR Medicine
The FDA issued draft guidance standardizing how genome editing therapies must be tested for off-target effects using next-generation sequencing -- the regulatory infrastructure for an emerging category of personalized CRISPR treatments for ultra-rare diseases.
The FDA issued draft guidance Monday establishing for the first time how companies developing genome editing therapies should use next-generation sequencing to assess whether their products cut DNA in unintended places. The guidance lays the regulatory groundwork for what could become an entirely new category of medicine: individualized CRISPR therapies designed for patients with ultra-rare genetic diseases.
What the Guidance Does
The document, titled "Safety Assessment of Genome Editing in Human Gene Therapy Products Using Next-Generation Sequencing," provides specific recommendations on sequencing strategies, sample selection, analysis parameters, and reporting for evaluating off-target editing risks.
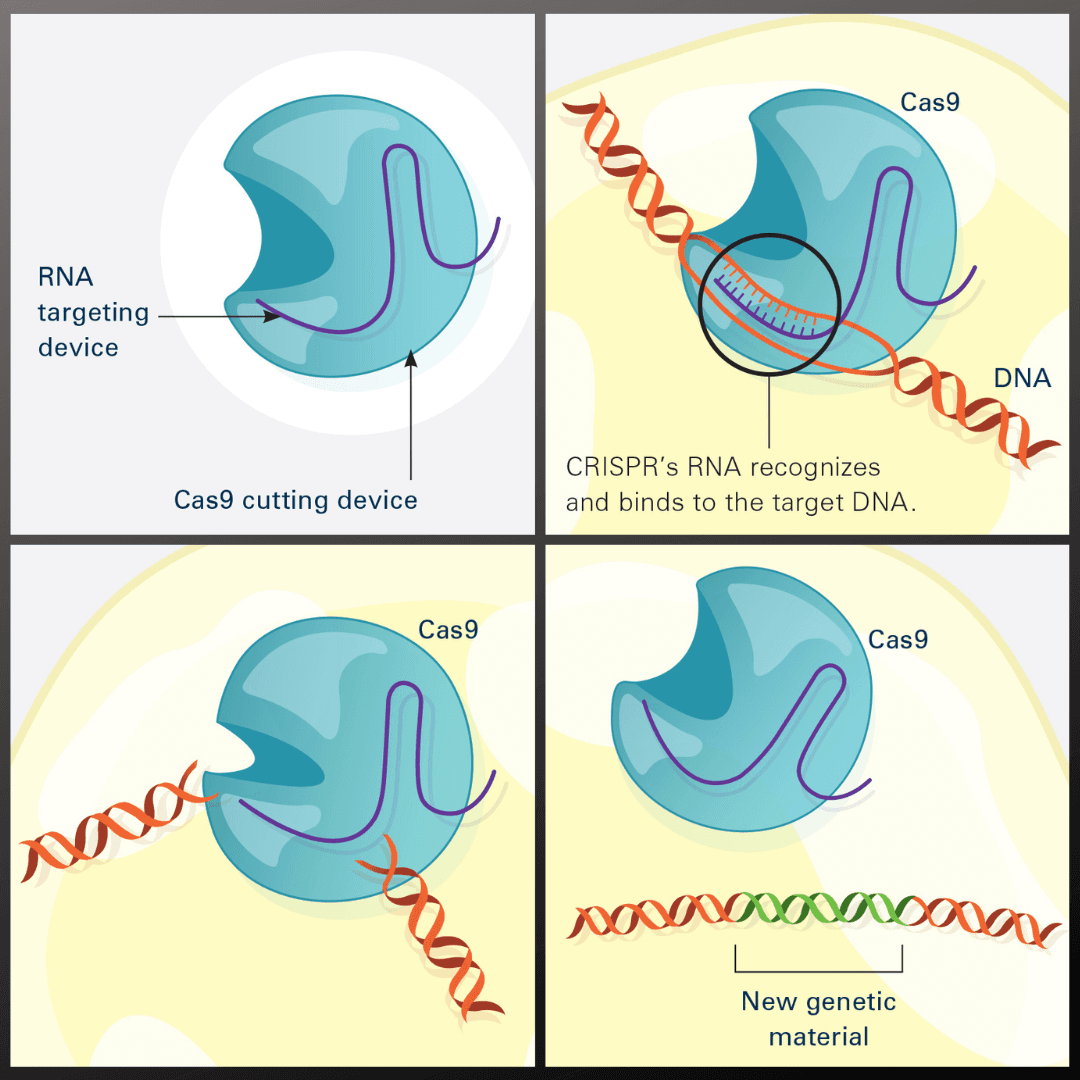
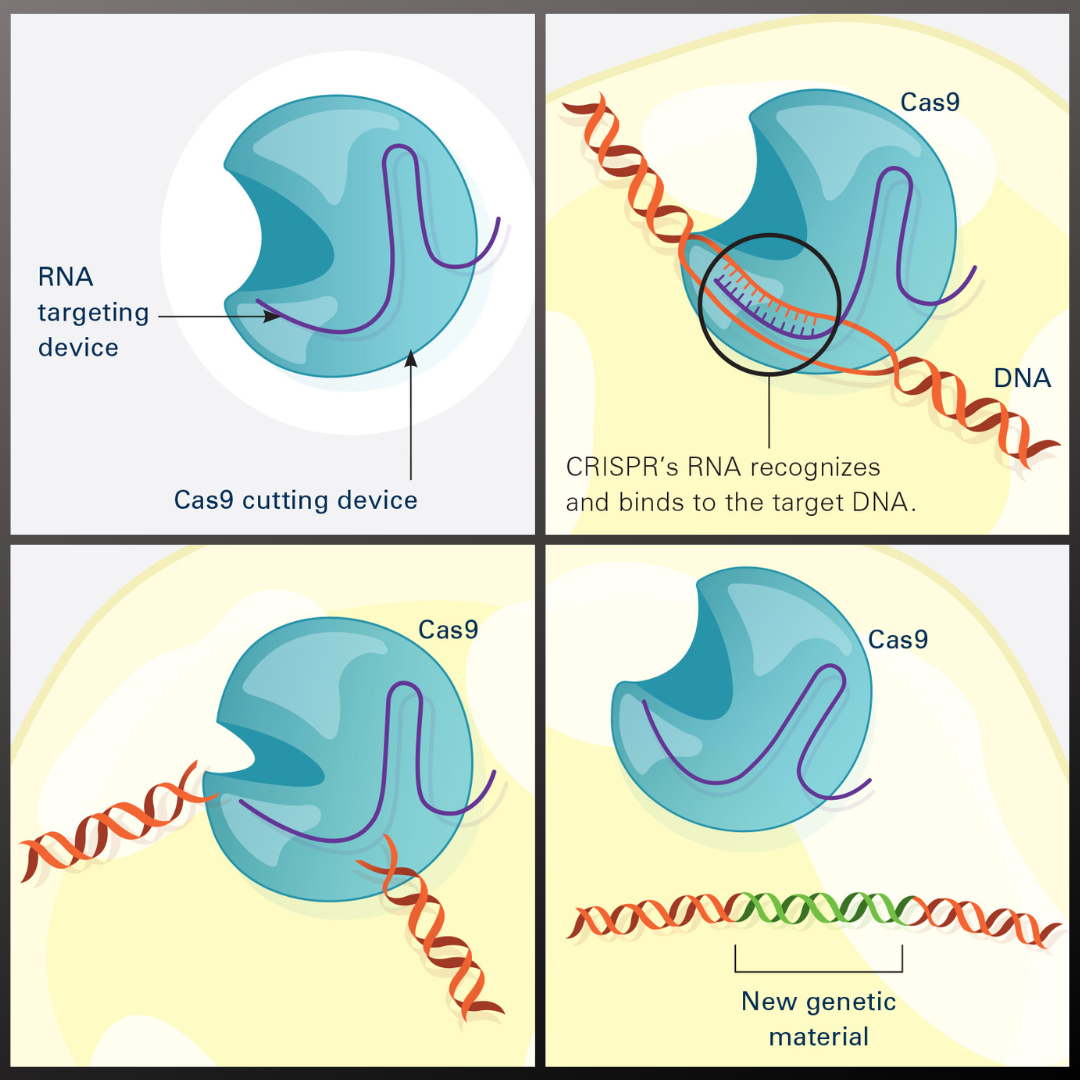
Genome editing therapies work by using molecular tools -- most commonly CRISPR-Cas9 -- to cut DNA at specific locations, either disabling a disease-causing gene or inserting corrective genetic material. The central safety concern is that these molecular scissors can sometimes cut at the wrong place in the genome, potentially causing unintended mutations. Next-generation sequencing can detect these off-target edits at high resolution across the entire genome.
The guidance applies to both categories of gene editing products:
- Ex vivo therapies, where a patient's cells are removed, edited in a laboratory, and returned to the body (the approach used in approved sickle cell treatments)
- In vivo therapies, where gene editing occurs directly inside the patient's tissues via an injected delivery vehicle
The recommendations cover nonclinical studies submitted with investigational new drug (IND) applications and Biologics License Applications (BLAs) -- the regulatory filings that determine whether a therapy can enter clinical trials and, ultimately, reach patients.
The Bigger Picture: Bespoke Therapies
The safety guidance is part of a broader initiative the FDA has been building since late 2025. In February, the agency launched the "plausible mechanism" framework -- a new regulatory pathway for individualized therapies targeting ultra-rare diseases where traditional randomized controlled trials are not feasible because so few patients exist.
The framework was inspired in part by the case of Baby KJ, a child with carbamoyl phosphate synthetase 1 (CPS1) deficiency who received the first personalized CRISPR therapy in 2025. The treatment used a base editor delivered via lipid nanoparticles to correct his specific mutation. After three infusions, he showed improved protein tolerance and ammonia control with no serious side effects.
The "plausible mechanism" pathway allows the FDA to grant marketing approval for therapies that demonstrate a scientifically credible mechanism of action, even without large clinical trials. Because genome editing therapies can be designed for specific mutations within the same gene, multiple variants of a therapy can be included under a single regulatory application using master protocols -- dramatically improving development efficiency.
Why Off-Target Detection Matters
The technical challenge the guidance addresses is fundamental: genome editing tools are not perfectly precise. A CRISPR guide RNA designed to target one location in the genome's 3 billion base pairs may also bind to similar sequences elsewhere, directing the Cas9 enzyme to make unintended cuts.
These off-target edits are rare in well-designed therapies but potentially dangerous -- they could disrupt essential genes, activate cancer-causing oncogenes, or cause chromosomal rearrangements. As individualized therapies proliferate, each with a unique guide RNA targeting a patient-specific mutation, the need for standardized safety assessment becomes acute. A one-off therapy designed for a single patient cannot be tested in hundreds of people first.
Next-generation sequencing provides the technological answer: it can survey the entire genome at sufficient depth to detect even low-frequency off-target events. The FDA's guidance standardizes how this sequencing should be performed, analyzed, and reported -- turning what has been an ad hoc process at individual labs into a uniform regulatory requirement.
"This guidance provides sponsors with clear, scientifically-grounded recommendations for evaluating off-target editing risks using state-of-the-art sequencing technologies," said FDA Commissioner Marty Makary.
Vinay Prasad, director of the Center for Biologics Evaluation and Research (CBER), said the guidance gives sponsors "a roadmap for comprehensive safety assessment while supporting the efficient development of these promising therapies."
What's Next
The draft guidance is open for public comment under docket number FDA-2026-D-1255. Once finalized, it will become the standard against which the FDA evaluates the safety data for all genome editing therapies seeking approval.
The guidance builds on the agency's January 2024 guidance on human gene therapy products incorporating genome editing, and complements the February 2026 bespoke therapy framework. Together, these documents form a regulatory architecture designed to move personalized genetic medicine from proof-of-concept to clinical practice.
The full draft guidance is available on the FDA website.